生物制品设计的基本要求GMP研讨会
admin 2019-12-23 09:52:19 316阅读
生物制品设计的基本要求GMP研讨会
1、生物制品
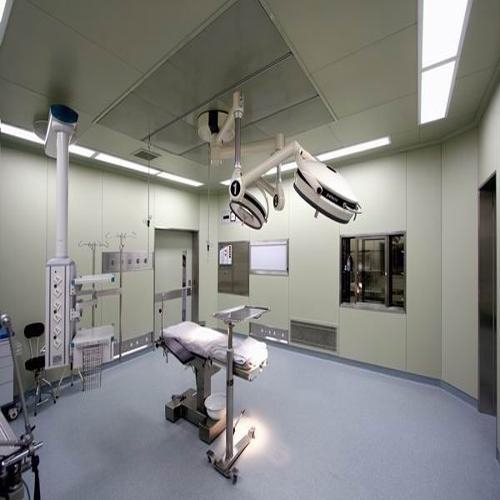
主要参考欧盟和世界卫生组织的相关GMP标准和2005年在中国修订的生物产品附录草案草案;重点是严格控制生产过程和中间过程以及一系列防止污染和交叉污染的要求:有毒作业区应有独立的空调系统;来自病原体操作区域的空气不应循环;来自第二类或更高病原体操作区域的空气应通过灭菌过滤器排出,并定期检查过滤器的性能;管理,特别是种子批次,细胞库系统(原始,主要代,工作)的管理要求以及生产操作和原材料的具体要求。成都手术室净化公司确保手术台洁净度达标,一般多采用垂直层流式效果较好 层流手术室是一个"正压"环境,其空气压力根据其不同区域(如手术间、无菌准备间、刷手间、麻醉间和周围干净区域等)洁净度不同要求而不同。成都十大黄色软件装修公司高效安全的手术室空气净化系统,保证手术室的无菌环境,可以满足器官移植、心脏、血管、人工关节置换等手术所需的高度无菌环境。采用高效低毒消毒剂,以及合理使用,是保障一般手术室无菌环境的有力措施。成都手术室装修公司采用空气洁净技术对微生物污染采取程度不同的控制,达到控制空间环境中空气洁净度适于各类手术之要求;并提供适宜的温、湿度,创造一个洁净舒适的手术空间环境。由于手术室要严格控制低细菌数及低麻醉气体浓度,所以层流超净装置的稳定性是层流净化手术室的重要验收指标。
2、血液制品
参考欧盟相关GMP附录、我国相关法律法规、“药典”标准、2007年血液制品生产整顿实施计划,关键内容是确保血液制品原料血浆、中间产品和成品的安全。它涉及原始血浆的再检验和检疫期、供体信息和产品信息的可追溯性、中间产品和成品安全指标的检查、检验、喂养生产、病毒灭活、不合格血浆处理等体外诊断试剂的管理等。
3、无菌药品
为确保无菌药物的安全性和质量提供监管和科学依据;采用欧盟,世卫组织,A,B,C,D等级标准;对无菌药物生产的清洁度提出了非常具体的要求。 ,动态监测;监测浮游细菌,沉淀细菌和表面微生物;完善中型模拟灌装,灭菌验证和管理的要求;增加无菌操作的具体要求;加强无菌保障措施。
药品GMP车间工艺布置应合理、紧凑,有利于生产操作,保证生产过程的有效管理。工艺布置应防止人流与物流的混合和交叉污染,并满足以下基本要求:
(一)设置进出生产区的客流通道,必要时设置特殊的出入口,如原材料、辅料(如原料、辅料、生产废弃物等)。清洁车间的传动路线应尽可能短。
2.进入清洁生产区域的人员和材料应有自己的净化室和设计。净化室的设定要求与生产区域的空气洁净度水平相适应。
三。生产作业区只设置必要的工艺设备和设计。用于生产、储存的区域不得作为非职工进入的通道。
4.运输人员和物资的电梯应分开。电梯不应位于清洁区域。必要时,应在电梯前安装锁阀室或其他措施,以确保清洁区域的空气清洁。
 检验科医学十大黄色软件区域装修施工技术要求
检验科医学十大黄色软件区域装修施工技术要求 四川成都洁净十大黄色软件装修公司哪家专业18980800355
四川成都洁净十大黄色软件装修公司哪家专业18980800355